Misoprostol is a synthetic analog of prostaglandin E1 (PGE1) developed for medical use, primarily in gastroenterology and obstetrics. It is designed to mimic the physiological actions of endogenous prostaglandins while exhibiting improved stability and oral bioavailability compared with naturally occurring prostaglandin compounds, which are rapidly degraded in vivo.
Prostaglandins are lipid-derived signaling molecules produced from arachidonic acid via the cyclooxygenase pathway. They function as local mediators involved in inflammation, smooth muscle activity, vascular tone, and protection of gastric mucosa. Prostaglandin E1, in particular, contributes to the regulation of gastric acid secretion and mucosal defense mechanisms.
Misoprostol is a methyl ester prodrug. After administration, it is rapidly de-esterified in the body to produce the biologically active free acid form, misoprostol acid. This active metabolite is responsible for the pharmacological effects of the drug. The prodrug design enhances absorption when administered orally and improves chemical stability compared with endogenous prostaglandins.
One of the primary clinical applications of misoprostol is the prevention of gastric ulcers associated with the use of nonsteroidal anti-inflammatory drugs (NSAIDs). NSAIDs inhibit cyclooxygenase enzymes, leading to reduced prostaglandin synthesis in the gastric mucosa. This reduction compromises mucosal protection, increasing susceptibility to ulceration. Misoprostol counteracts this effect by activating prostaglandin receptors that promote mucus and bicarbonate secretion, increase mucosal blood flow, and reduce gastric acid secretion.
In addition to its gastrointestinal use, misoprostol has important applications in obstetrics and gynecology. It induces uterine contractions by activating prostaglandin receptors in uterine smooth muscle. This property is utilized in medical management of miscarriage, induction of labor, and medical termination of pregnancy in appropriate clinical settings. The uterotonic effect is mediated through increased intracellular calcium levels in smooth muscle cells following receptor activation.
Misoprostol exerts its biological activity through binding to prostaglandin E receptors (EP receptors), which are G protein–coupled receptors with multiple subtypes (EP1, EP2, EP3, and EP4). The specific physiological response depends on receptor subtype distribution in target tissues. In the stomach, EP3 receptor activation is particularly important for reducing acid secretion, while in uterine tissue, multiple EP receptors contribute to contractile responses.
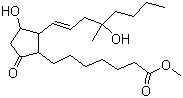
From a structural perspective, misoprostol is a lipid-like molecule derived from the prostaglandin E1 scaffold. It contains a cyclopentane ring with multiple stereocenters and two aliphatic side chains. The stereochemistry of these centers is essential for receptor binding and biological activity, as prostaglandin receptors are highly stereospecific.
The drug is highly lipophilic in its esterified form, which facilitates gastrointestinal absorption. After conversion to misoprostol acid, the molecule becomes more polar and interacts effectively with prostaglandin receptors in target tissues. This balance between lipophilicity for absorption and polarity for receptor activity is a key feature of prostaglandin prodrug design.
Misoprostol is rapidly metabolized and has a relatively short half-life, which contributes to its transient but effective pharmacological action. Its safety and efficacy profile depend on dose and route of administration, and its effects on smooth muscle and gastric secretion are dose-dependent.
Overall, misoprostol is a synthetic prostaglandin E1 analog that functions as a prodrug for misoprostol acid. It acts on EP prostaglandin receptors to reduce gastric acid secretion and stimulate uterine contractions, making it clinically important in both gastroprotective therapy and obstetric applications. Its significance lies in its dual pharmacological roles and its representation of successful prostaglandin analog design for therapeutic use.
References
2026. Gastroprotective effect of salvianolic acid B on rat model of diclofenac-induced gastric ulceration. Comparative Clinical Pathology.
DOI: 10.1007/s00580-026-03758-2
2026. A comparative study of some NSAIDs with natural products: integrating in vitro anticancer efficacy, in vivo antiulcerative effect, histochemistry, and in silico analysis. Naunyn-Schmiedeberg's Archives of Pharmacology.
URL: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC13152882
|







 GHS06;GHS08 Danger Details
GHS06;GHS08 Danger Details chemBlink Chemical Story
chemBlink Chemical Story